

ОПРЕДЕЛЕНИЕ
Наследственное заболевание, в основе которого лежит нарушение экскреции желчных кислот через канальцевую мембрану гепатоцита. Впервые описано у детей, потомков Джакоба Байлера, и с тех пор названо его именем.
КОД ПО МКБК76.8 - Другие уточненные болезни печени.
ЭТИОЛОГИЯ
Прогрессирующий семейный внутрипеченочный холестаз - следствие генетически-детерминированного нарушения структуры канальцевой мембраны гепатоцита. Имеет аутосомно-рецессивный тип наследования и включает три основных типа: 1тип (болезнь Байлера), II тип (синдром Байлера), III тип (дефицит MDR3 гена). В основе I и II типа лежит нарушение экскреции желчных кислот, тогда как III тип обусловлен нарушением экскреции фосфолипидов.
Ген, ответственный за развитие заболевания, локализуется в регионе длинного плеча 18-й хромосомы (18q21), протяженностью 7сМ в интервале между маркерами D18S69 и D18S64.
ПАТОГЕНЕЗ
В основе болезни Байлера лежит дефицит мембрана-связанного фермента - П-типа АТФ-азы, играющего ключевую роль в транспорте жирорастворимых соединений и желчных кислот через канальцевую мембрану гепатоцита. Вследствие этого дефекта первичные желчные кислоты накапливаются в клетках печени и оказывают на них повреждающее действие, способствуя их разрушению (пусковой фактор апоптоза). С другой стороны, первичные желчные кислоты не поступают в желчную систему и, следовательно, в кишечник, что приводит к нарушению процессов всасывания жиров и жирорастворимых витаминов.
КЛИНИЧЕСКАЯ КАРТИНА
Появление первых признаков холестаза в большинстве случаев отмечают в период новорожденнсти, реже в возрасте 1-10 месяцев жизни. Описаны также случаи желудочно-кишечных и внутричерепных кровотечений, которые предшествовали появлению других клинических признаков болезни. Характерна желтуха, умеренно выраженная гепатомегалия, непостоянная ахолия стула и темный цвет мочи. Типичный признак болезни Байлера - кожный зуд, появляющийся уже в течение первых трех месяцев жизни. Отставание ребенка в физическом развитии и наличие признаков дефицита жирорастворимых витаминов (рахитические изменения и остеопения, мышечная гипотония, сухость кожи и слизистых, тусклость и ломкость ногтей и волос, офтальмоплегия, петехиальная сыпь и/или кровоточивость слизистых) - также характерны для данного заболевания. Синдром холестаза при болезни Байлера имеет волнообразное течение. Факторами, способствующими нарастанию клинико-лабораторных признаков холестаза, служат инфекционные заболевания верхних дыхательных путей и другие интеркурентные заболевания.
ДИАГНОСТИКА
Пренатальиая
Указанные генетические маркеры 18-й хромосомы могут быть также использованы для пренатальной диагностики заболевания и генетического консультирования.
Физикальное исследование
Необходимо оценить цвет кожного покрова и склер, размеры печени и селезенки, цвет стула и мочи. У детей старше трех месяцев жизни может быть кожный зуд.
Лабораторные исследования
Низкий уровень ГГТ и холестерина сыворотки крови наряду с повышением других маркеров холестаза, и в том числе ЩФ, прямой фракции билирубина и желчных кислот.
Характерно повышение ферментов цитолиза и отсутствие изменений белок-синтетической функции печени.
Часто отмечают удлинение ПВ или снижение протромбинового индекса, генез которого связан с нарушением всасывания витамина К в кишечнике.
Для уточнения диагноза при болезни Байлера возможно проведение молекулярно-генетического тестирования специфического локуса длинного плеча 18-й хромосомы (18q21), протяженностью 7сМ в интервале между маркерами D18S69 и D18S64.
Инструментальные исследования
При проведении пункционной биопсии печени отмечают наличие преимущественно внутриклеточного холестаза. Вторично наступает перегруппировка гепатоцитов, образующих тубулярные структуры, псевдоканальца и формирование билирного цирроза печени. Электронная микроскопия обнаруживает желчь в виде грубых гранул («желчь Байлера») в гепатоцитах и внутрипеченочных желчных канальцах.
Дифференциальная диагностика
Проводят с другими заболеваниями печени, проявляющимися внутрипеченочным холестазом с низким уровнем фермента ГГТ (синдром Цельвейгера, нарушение синтеза желчных кислот вследствие ферментопатии).
Показана консультация клинического генетика.
ЛЕЧЕНИЕ
Цели лечения
Коррекция осложнений длительно сохраняющегося холестаза.
Немедикаментозное лечение
Медикаментозное лечение
Урсодезоксихолевая кислота в дозе 20-30 мг/(кгхсут) в 2 приема - постоянно. Жирорастворимые витамины, макро- и микроэлементы (см. лечение АВЖП). У детей более старшего возраста при развитии кожного зуда используют следующие препараты: холестерамин в дозе 4-16 г/день, рифампицин 10 мг/кгхсут) и другие, фототерапия, плазмоферез. Используют также средства, воздействующие на рецепторный аппарат кожи, такие, как ментоловое масло, ланолин, теплые ванны и т.д.
Хирургическое лечение
При развитии патологических состояний, нарушающих качество жизни больного (кожный зуд, отставание в физическом развитии, изменения, обусловленные дефицитом жирорастворимых витаминов), проводят трансплантацию печени.
Дальнейшее ведение
Медикаментозное лечение проводят постоянно. Динамическое амбулаторное обследование 1 раз в 1-2 месяца или по показаниям.
Без трансплантации печени прогноз заболевания неблагоприятный. Больные умирают в возрасте от 2 до 15 лет. Описаны отдельные больные с продолжительностью жизни до 25 лет. Кроме того, по мере прогрессирования заболевания возможно развитие рака печени и желчевыводящей системы.
Прогрессирующий семейный внутрипеченочный холестаз II типа (синдром Байлера)
ОПРЕДЕЛЕНИЕ
Наследственное заболевание, в основе которого лежит нарушение экскреции преимущественно одной первичной желчной кислоты (хенодезоксихолевой), через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности П-гликопротеина.
КОД ПО МКБ-К76.8 - другие уточненные болезни печени.
ЭПИДЕМИОЛОГИЯ
Заболевание встречается в изолированных популяциях на Среднем Востоке. Гренландии и Швеции.
ЭТИОЛОГИЯ
Ген, ответственный за развитие данного заболевания, локализуется во 2-й хромосоме (2q24). Этот ген имеет молекулярную структуру, похожую на таковую гена, ответственного за развитие I типа прогрессирующего семейного внутрипеченочного холестаза, в связи с чем его обозначают «сестринским». В основе прогрессирующего семейного внутрипеченочного холестаза II типа лежит нарушение экскреции преимущественно хенодезоксихолевой кислоты через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности П-гликопротеина.
ПАТОГЕНЕЗ
Не отличается от 1 типа прогрессирующего семейного внутрипеченочного холестаза.
КЛИНИЧЕСКАЯ КАРТИНА
ДИАГНОСТИКА
Пренатальная
Указанные генетические маркеры 2-й хромосомы могут быть использованы для пренатальной диагностики заболевания и генетического консультирования.
Физикальное исследование
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Лабораторные исследования
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Дифференциальная диагностика
Проводят с другими заболеваниями печени, проявляющимися внутрипеченочным холестазом с низким уровнем фермента ГГТ. Показана консультация клинического генетика.
Этиопатогенетическое.
Цели лечения: коррекция осложнений длительно сохраняющегося холестаза.
Немедикаментозное лечение
Лечебное питание с повышенным содержанием среднецепочечных триглицеридов.
Медикаментозное лечение
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Хирургическое лечение
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Дальнейшее ведение
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Патофизиологические основы формирования
неонатального холестаза
Мухина Ю.Г, Дегтярева А.В., Морозов И.А, Туманова Е.Л, Талалаев А.Г.
Кафедра детских болезней № 2 педиатрического факультета с курсом гастроэнтерологии и диетологии ФУВ, Кафедра патологической анатомии педиатрического факультета, РГМУ, ЦНИИГ.
Холестаз – нарушение образования и/или экскреции желчи по желчевыводящей системе, приводящее к внутриклеточному и/или внутрипротоковому накоплению компонентов желчи и проявляющееся желтухой, постоянной или периодической ахолией стула, темным цветом мочи, увеличением размеров печени, кожным зудом, повышением уровня прямой фракции билирубина, щелочной фосфатазы (ЩФ), гамма-глутаминтрансферазы (ГГТ), холестерина, бета-липопротеидов (b -ЛПД) и желчных кислот (ЖК).
В норме секреция желчи зависит от активности ферментных и транспортных систем гепатоцитов и желчных протоков, а также от их структурного и функционального их взаимодействия. Образование первичных ЖК (холевой и хенодезоксихолевой) происходит исключительно в печени из холестерина. Образование вторичных ЖК – дезоксихолевой и литохолевой происходит вследствие 7 a -дегидроксилирования первичных под действием бактерий кишечника. Третичные ЖК и, в основном, урсодезоксихолевая синтезируются в печени путем изомеризации вторичных ЖК. Печеночно-кишечная циркуляция ЖК включает транспорт ЖК в крови, их захват гепатоцитом, транспорт внутри гепатоцита от синусоидального (базолатерального) полюса к канальцевому, конъюгацию деконъюгированных в кишечнике и ре-синтезированных ЖК, экскрецию их в кишечник и последующую реабсорбцию как конъюгированных, так и неконъюгированных ЖК.
Экскреторная функция является жизненно важной; нарушение которой приводит не только к патологическим изменениям гепатобилиарной системы, но и вовлекает в патологический процесс другие органы и системы. Синдром холестаза способствует реализации токсических эффектов различных компонентов желчи, из которых первостепенное значение имеют желчные кислоты, билирубин, холестерин, провоспалительные медиаторы и микроэлементы. Токсические эффекты компонентов желчи представлены в табл. 1.
Таблица 1.
Патологические эффекты компонентов желчи при синдроме холестаза.
|
Компоненты желчи: |
Патологические эффекты: |
|
Желчные кислоты |
Цитотоксичность, нарушение дыхательной цепи митохондрий, апоптоз, повреждение канальцев. |
|
Билирубин |
Нарушение структуры митохондрий |
|
Холестерин |
Повреждение клеточных мембран |
|
Лейкотриены |
Воспаление, гемодинамические эффекты |
|
Медь |
Перекисное окисление липидов |
Дефицит желчи в кишечнике способствует дисрегуляции желудочно-кишечного тракта, нарушению процессов всасывания и, в первую очередь, жиров и жирорастворимых витаминов, нарушению функционального состояния поджелудочной железы. Отставание детей в физическом развитии является характерным признаком хронических холестатических заболеваний гепатобилиарной системы. Вторично формирующийся дисбаланс микроэлементов нарушает процессы оссификации костной системы с развитием остеопороза и метаболических нарушений сердечно-сосудистой системы. Например, при синдроме Алажиля длительно в течение нескольких лет существующий синдром холестаза с постоянно высоким уровнем холестерина и других липидов в крови способствует атеросклеротическим изменениям сосудов уже в детском возрасте. Высокий уровень сывороточных желчных кислот является ведущим фактором в развитии кожного зуда. Основные внепеченочные проявления длительно сохраняющегося синдрома холестаза представлены в таблице 2. Крайний вариант – на фотографии.
Таблица 2.
Внепеченочные проявления синдрома холестаза, фотография ребенка М.
|
Нарушение процессов всасывания |
|
|
Отставание в физическом развитии |
|
|
Признаки дефицита жирорастворимых витаминов: Остеопороз Нейропатия Ксерофтальмия Коагулопатия |
|
|
Кожный зуд |
|
|
Ксантомы, атеросклеротические изменения сосудов. |
У новорожденных и детей первых месяцев жизни синдром холестаза является одним из ранних признаков широкого спектра заболеваний печени и желчевыводящих протоков. В генезе заболеваний лежит несостоятельность тех или иных структур гепатобилиарной системы, являющихся неотъемлемым звеном сложной системы образования и экскреции желчи. Эти дефекты могут быть генетически детерминированными или возникать под действиемэкзогенных факторов. В таблице 3 представлены заболевания гепатобилиарной системы в зависимости от уровня первичного поражения.
Таблица 3.

Первичное поражение клеток печени характерно для большинства гепатотропных инфекционных возбудителей, кроме того может наблюдаться при эндокринных и метаболических нарушениях, а также как проявление токсического действия лекарственных препаратов и осложнение длительного полного парентерального питания.
Гепатит,протекающий с синдромом холестаза, у новорожденных детей может быть вызван широким спектром инфекционных агентов: герпес-вирусом, цитомегаловирусом, энтеровирусами, вирусом краснухи, Эпштейна-Барра, гепатита В и С, возбудителем токсоплазмоза, листериоза, сифилиса и бактериальными агентами. В большинстве случаев неонатальный гепатит, вызванный вышеуказанными возбудителями, является одним из проявлений генерализованной инфекции. Выявление признаков инфекционного процесса и характерного для той или иной инфекции симптомокомплекса является необходимым условием для диагностики гепатита. Исключение составляют вирусные гепатиты В и С, при которых могут отмечаться изменения только печени. Однако у новорожденных детей гепатиты В и С встречаются редко.
Механизм патологического действия включает непосредственное повреждающее действие гепатотропных возбудителей с развитием специфического воспаления гепатобилиарной системы. При неонатальном гепатите синдром холестаза сочетается с биохимическим синдромом цитолиза и нарушением белок-синтетической функции печени. Характерно повышение неспецифических показателей воспаления в общем и биохимическом анализах крови. Наиболее объективным доказательством неонатального гепатита являются результаты морфологического исследования биоптата печени, при котором выявляются типичные для каждой инфекции признаки. Исследования, выявляющие возбудителя и/или антитела к нему подтверждают диагноз.
В настоящее время особенный интерес представляет ЦМВ инфекция. Инфицирование плода на ранних сроках внутриутробного развития может быть одной из причин формирования пороков и/или аномалий развития, в том числе пороков желчевыводящей системы в виде атрезии внепеченочных желчных протоков или гипоплазии внутрипеченочных желчных протоков. При этом ребенок рождается с признаками врожденных пороков и не имеет проявлений гепатита. Нередко встречаются бессимптомные варианты течения, при которых отсутствуют изменения гепатобилиарной системы. В 10-15% случаев клинически не выраженной инфекции могут развиваться поздние проявления, такие как сенсорная глухота, трудности в обучении, а также минимальные мозговые дисфункции. Синдром острой врожденной ЦМВИ (цитомегалия, инклюзионная болезнь) встречается редко и является следствием инфицирования на поздних сроках внутриутробного развития. Типичными для данного синдрома являются: низкая масса тела при рождении, геморрагическая сыпь, тромбоцитопения, анемия, желтуха, гепатоспленомегалия, микроцефалия, нефрит и хориоретинит. Развитие специфического гепатита при этой форме отмечается менее чем в 50% случаев, тогда как чаще выявляется неспецифическая реакция со стороны печени. Диагноз устанавливается на основании выше перечисленного симптомокомплекса, клинико-биохимических признаков гепатита, а также результатов пункционной биопсии печени. Патогномоничным гистологическим признаком гепатита ЦМВ-этиологии является выявление гигантских клеток с ЦМВ включениями - клеток «совиный глаз» (рис. 1).

Рис. 1. Гистологическое исследование биоптата печени при ЦМВ-гепатите.
Среди метаболических заболеваний манифестирующих в течении первых месяцев жизни и проявляющихся синдромом холестаза следует отметить галактоземию, фруктоземию, тирозинемию, болезнь Нимана-Пика (тип С), дефицит-a -1-антитрипсина, неонатальный гемохроматоз и некоторые типы митохондриальной недостаточности с вовлечением ферментов цепи переноса электронов. В основе этих нарушений лежит генетически обусловленный дефект ферментной системы, приводящий к накоплению существующих в норме соединений, а также промежуточных продуктов их метаболизма, обладающих токсическим эффектом на клетки различных органов и являющихся ключевым патогенетическим фактором.
Наиболее часто встречается галактоземия, которая является наследственным заболеванием с аутосомно-рецессивным типом наследования, связанным с 9 хромосомой и включает патологические изменения со стороны печени, почек, ЦНС и глаз. Это заболевание встречается с частотой 1: 50.000 (1:18.000 -1:180.000) живорожденных новорожденных. Первые признаки заболевания появляются в период от нескольких часов до нескольких дней после начала энтерального питания молоком или смесью, содержащей галактозу. В основе этого заболевания лежит дефицит фермента галактоза-1-фосфатуридилтрансферазы, приводящий к накоплению галактозы и галактоза-1-фосфата. Галактоза превращается в галактитол, ответственный за развитие катаракты. Галактоза-1-фосфат значительно снижает синтез энергетических соединений (АТФ, ГТФ, ЦТВ), а также активность ферментов, принимающих участие в глюконеогенезе и синтезе глюкозы из гликогена, вызывая гипогликемию. Эти изменения вызывают тяжелые метаболические нарушения в клетках печени, головного мозга, почек и глаз, способствуют гемолизу эритроцитов. Галактоза-1-фосфат превращается в галактонат и галактонолактон. Эти метаболиты обладают непосредственным гепато-, нефро- и нейротоксическим действием. Описанные изменения лежат в основе характерного для галактоземии симптомокомплекса в виде нарушения общего состояния ребенка, проявляющегося срыгиваниями, рвотой, расстройством стула и др., признаками гемолитической анемии, патологическими изменениями со стороны печени в виде синдрома холестаза, синдрома цитолиза и нарушения синтетической функции. Первым клиническим признаком изменений со стороны ЦНС является внутричерепная гипертензия. Отмечаются также нарушения канальцевой функции почек и формирование катаракты. Подтверждением диагноза служит высокий уровень галактозы в крови, а также дефицит фермента галактоза-1-фосфатуридилтрансферазы.
Характерной особенностью вышеперечисленных метаболических заболеваний также являются патологические изменения со стороны различных органов. Внепеченочные признаки часто предшествуют клинико-лабораторным проявлениям холестаза. Во всех случаях выявляются патологические изменения ЦНС, почек и глаз. У этих больных отмечаются раздражительность, выраженные срыгивания и рвота, плохая прибавка массы тела, частый жидкий стул, клинические эквиваленты гипогликемии и ряд других признаков. При неонатальном гемохроматозе и митохондриальной недостаточности отмечается клиническая симптоматика полиорганной недостаточности. При фруктоземии существует связь между введением в рацион продуктов, содержащих фруктозу или сахарозу и появлением первых признаков заболевания. Особенностью тирозинемии является «капустный запах» и фотофобия, связанная с развитием кератита. При метаболических нарушениях, так же как и, при инфекционных заболеваниях, существует характерный для каждого заболевания симптомокомплекс, играющий ведущую роль в диагностике. Результаты биопсии печени, а также специфические исследования, выявляющие соответствующие дефекты, подтверждают диагноз.
Гистологические признаки имеют особое значение при болезни Нимана-Пика (тип С) и при неонатальном гемохроматозе. При болезни Нимана-Пика типе С вследствие дефицита фермента кислой сфингомиелиназы, участвующей в эстерификации эндогенного холестерина отмечается его накопление в лизосомах макрофагов, что оказывает на них повреждающее действие. При гистологическом исследовании макрофаги, переполненные липидами, выглядят в виде «пенистых» клеток (рис. 2). При неонатальном гемохроматозе диагностическое значение имеет накопление железа в клетках печени, тогда как в норме накопление железа осуществляется в Купферовских клетках.

Рис. 2. Гистологическое исследование биоптата печени при болезни Нимана-Пика, тип С.
Дефицит-a -1-антитрипсина отличается от описанных метаболических нарушений. Единственным его проявлением в неонатальном периоде является синдром холестаза; общее состояние ребенка, как правило, не страдает, патологические изменения в легких развиваются значительно позже, после 3-5 лет жизни. Заболевание имеет аутосомно-рецессивный тип наследования, связанный с нарушением одной аминокислотной цепочки гена, локализующегося в 14 хромосоме.
Конформационные изменения a 1-антитрипсина (a 1-АТ) обуславливают его соединение с соответствующими ферментами. В связи с этим, те или иные структурные нарушения способствуют ослаблению или потере связывающей способности. Фенотипические варианты a 1-АТ отражают скорость его миграции в электрическом поле при pH между 4 и 5. Нормальный белок мигрирует в промежуточной, средней позиции и обозначается М (medium ), белок, мигрирующий более медленно, обозначают Z или S (slow ). В редких случаях отмечается нулевой фенотип - null , при котором полностью отсутствует белок. Наличие двух пар генов предполагает 6 основных вариантов фенотипа, отличающихся степенью выраженности дефицита: MM -100%, MS -75%, MZ -57%, SS -52%, SZ -37%, ZZ -16%. При Z -варианте отмечается замена глутамина на лизин в 342 положении, при S варианте глутамина на валин в 264 положении. Кроме того, при ZZ типе наряду с изменением аминокислотной последовательности имеет место нарушение эндоплазматического ретикулума гепатоцитов, что приводит к нарушению образования боковых углеводородных цепей. В связи с этим только 15% синтезированных полипептидов секретируется в плазму крови, тогда как 85% откладывается в эндоплазматическом ретикулуме, образуя глобулярные включения. Накопление белковых гранул при ZZ фенотипе является ведущим патогенетическим фактором патологических изменений печени. При S варианте отсутствуют патологические изменения гепатобилиарной системы, a -1-АТ не накапливается в клетках печени, однако он имеет нестабильную структуру и подвергается частичному внутриклеточному протеолизу, что обуславливает его сывороточный дефицит. Примерно в 21-50% случаев при неонатальном холестазе, обусловленном дефицитом a 1-АТ по ZZ фенотипу, отмечается развитие ювенильного цирроза печени. Наиболее типичным бронхолегочным проявлением заболевания, встречающимся при любом фенотипе у детей старше 3-5 летнего возраста, является эмфизема легких, однако дефицит a 1-АТ играет важную роль в патогенезе и других хронических заболеваний легких и, прежде всего бронхиальной астмы. Ориентировочным тестом, свидетельствующим о дефиците a -1-антитрипсина, является низкий сывороточный уровень a -1-глобулина. Подтвердить диагноз позволяет низкий уровень a -1-АТ наряду с выявлением характерных гистологических признаков в виде ШИК положительных включений, устойчивых к ферментативному перевариванию и располагающихся преимущественно в перипортальных зонах (рис. 3).

Рис. 3. Гистологическое исследование биоптата печени придефиците a -1-АТ.
Канальцевая мембрана гепатоцита несет на своей поверхности транспортные системы, отвечающие за экскрецию различных компонентов желчи, органических и неорганических веществ, а также большинства ксенобиотиков. В генезе формирования синдрома холестаза играет роль генетически детерминированные дефекты 3-х транспортных систем канальцевой мембраны гепатоцита, лежащих в основе прогрессирующего семейного внутрипеченочного холестаза (ПСВХ) 3-х типов (табл. 4). Н аиболее часто встречается ПСВХ 1-го типа или болезнь Байлера, в основе которой лежит дефицит II -типа АТФ-азы, играющей ключевую роль в экскреции обоих первичных желчных кислот через канальцевую мембрану гепатоцита.При 11 типе ПСВХ отмечается нарушение экскреции преимущественно одной, хенодезоксихолевой кислоты через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности П-гликопротеина.
Таблица 4.
Типы и основные характеристики прогрессирующего семейного внутрипеченочного холестаза.
|
Типы: |
Генный дефект: |
Молекулярные изменения канальцевой мембраны гепатоцита: |
Начальный патогенетический дефект: |
Своеобразие маркеров холестаза: |
|
(болезнь Байлера) |
18q 21 |
Мутация II -типа |
Нарушение экскреции жирорастворимых соединений и в т.ч.желчных кислот |
Низкий уровень ГГТ и холестерина, Повышение ЩФ |
|
(синдром Байлера) |
2q24 |
Отсутствие II -гликопротеина |
Нарушение экскреции преимущественно хенодезоксихолевой кислоты |
Низкий уровень ГГТ и холестерина Повышение ЩФ |
|
III (дефицит MDR -3 гена) |
7q 21.1 |
Отсутствие MDR 3 - II -гликопротеина |
Нарушение экскреции фосфолипидов |
Высокий уровень ГГТ и холестерина Повышение ЩФ |
Примечание: II -тип АТФ-азы - фермент, локализующийся на канальцевой мембране гепатоцита, ответственный за экскрецию преимущественно первичных желчных кислот.
II -гликопротеин - транспортный белок канальцевой мембраны гепатоцита, обеспечивающий
экскрецию, преимущественно хенодезоксихолевой кислоты.
MDR 3 - II -гликопротеин - транспортная система канальцевой мембраны гепатоцита, ответственная за экскрецию фосфолипидов и прежде всего фосфатидилхолина.
Вследствие этих дефектов первичные ЖК накапливаются в клетках печени, а по мере достижения определенной критической концентрации оказывают токсическое действие на гепатоциты и поступают в кровь, способствуя развитию кожного зуда. Неполный вариант синдрома холестаза, имеющий место при этих заболеваниях, проявляющийся нарушением экскреции только ЖК, значительно нарушает процессы кишечного всасывания. Клинические проявления включают синдром холестаза со значительной степенью выраженности кожного зуда, а также отставание детей в физическом развитии и признаки дефицита жирорастворимых витаминов.Характерной лабораторной особенностью I и II типов служит низкий уровень ГГТ и холестерина сыворотки крови наряду с повышением других маркеров холестаза. Фермент ГГТ является мембрано-связанным, локализующимся преимущественно во внутрипеченочных желчных протоках. Основным стимулом для его выделения служат ЖК и, следовательно, заболевания, при которых ЖК не поступают во внутрипеченочную систему, не будут сопровождаться повышением уровня данного фермента. Следует отметить, что в большинстве случаев низкий уровень ГГТ сыворотки крови сочетается с низким уровнем холестерина. Диагностическое значение имеет исследование ЖК, позволяющее определить высокий их уровень в крови и отсутствие или следовые концентрации в желчи.Морфологически выявляется преимущественно внутриклеточное скопление крупных гранул желчи – желчи Байлера (рис. 4).

Рис. 4. Электронно-микроскопическое исследование биоптата печени при болезни Байлера.
В основе III типа ПСВХ (дефицита MDR 3-гена) лежит нарушение экскреции фосфолипидов и, прежде всего фосфатидилхолина, через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности MDR 3-П-гликопротеина. Основным патогенетическим фактором является повреждение внутрипеченочных желчных протоков поступающими в их просвет свободными, несвязанными с фосфолипидами, желчными кислотами. Характерно вовлечение в патологический процесс мелких желчных капилляров, тогда как другие уровни желчевыводящей системы остаются интактными. При гистологическом исследовании определяется пролиферация желчных протоков с постепенным формированием фиброза. При ретроградной или чрезкожной холангиографии изменения не выявляются. Подтверждением диагноза является отсутствие фосфолипидов в желчи и повышенное их содержание в крови.
Важными структурными элементами гепатобилиарной системы являются межклеточные соединения, состоящие из эпителия двух типов: плотного и рыхлого, проницаемого для воды и растворов, обеспечивающие осмотическое равновесие между кровью и желчью, а также межклеточный обмен глюкозой, ионами и аминокислотами (рис. 5а). Межклеточные плотные контакты имеют сложную белковую структуру, схематическое изображение которых представлено на рис. 5б.

Рис. 5 (а). Межклеточные соединения (плотные контакты) в норме (электронная микроскопия)

Рис. 5 (б). Схематичное изображение м ежклеточных плотных контактов.
При развитии холестаза отмечается грубое, неселективное увеличение проницаемости межклеточных соединений, позволяющее осуществлять обратную диффузию канальцевого содержимого, что рассматривается как механизм “предохраняющий” гепатоцит от повреждения. С другой стороны генетически детерминированный дефект межклеточных соединений может быть причиной внутрипеченочного холестаза (рис. 6). Описанный дефект способствует повышенному содержанию компонентов желчи в крови, изменению коллоидных свойств желчи, что приводит к более длительному ее сохранению в клетках печени и внутрипеченочных желчных канальцах.

Рис. 6. Генетически-детерминированный дефект плотных контактов (электронная микроскопия)
Желчевыводящие протоки выполняют важную функцию окончательного формирования желчи, что достигается путем реабсорбции и секреции ее компонентов. В этом процессе задействованы различные транспортные системы и в том числе хлоридные каналы, ответственные за обмен ионов хлора и бикарбоната, а также натрия и водорода, генетический дефект которых лежит в основе муковисцидоза. Нарушение секреции воды, бикарбонатов и других веществ изменяет коллоидное состояние желчи, нарушает ее отток, приводит к длительной обструкции желчных протоков с развитием билиарного цирроза.
Эпителиальные клетки желчных протоков (холангиоциты) отвечают за секрецию биологически активных веществ – IL 6, TGF -β, NO , белка хемотаксиса моноцитов, эндотелина 1, обеспечивая тем самым взаимосвязь с клетками других органов и систем. Возможно, несостоятельность секреторной функции холангиоцитов наряду с генетическими и инфекционными факторами играет определенную роль в формировании хронического воспалительного процесса при первичном склерозирующем холангите. При этом заболевании вовлечение различных участков желчевыводящих путей неодинаковое. Оно может быть ограничено внутри- и внепеченочными желчными протоками. Диагностическими критериями являются четкообразные изменения и стеноз желчных путей, выявляемые при холангиографии (рис. 7а). Гистологически выявляется пролиферация желчных протоков с постепенным формированием вокруг протоков фиброза в виде луковичной шелухи, значительное отложение меди и ступенчатые некрозы (рис. 7б).

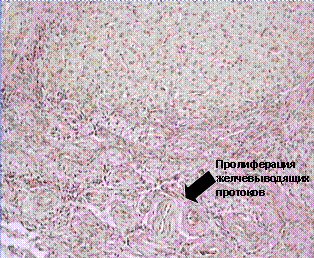
Рис. 7.(а, б). Ретроградная холангиография (а), гистологическое исследование биоптата печени (б) при первичном склерозирующем холангите.
Врожденная гипоплазия внутрипеченочных желчных протоков, проявляющаяся неонатальным холестазом, может сочетаться с аномалиями и/или пороками развития других органов и соответствовать синдрому Алажиля. Диагноз данного синдрома устанавливается на основании выявления двух и более аномалий со стороны других органов (табл. 5).
Таблица 5.
Частота встречаемости диагностических критериев синдрома Алажиля.
Авторы:Основные критерии: |
Emerick et al |
Alagille
|
Deprettere
|
Средний процент |
Синдром внутрипеченочного холестаза |
96% (88/92) |
100%(80/80) |
93% (25/27) |
96% |
|
ВПС (периферический стеноз или гипоплазия легочной артерии) |
||||
|
Офтальмологические изменения (задний эмбриотоксон) |
||||
|
Аномалии развития скелета (расщепление тел позвонков в виде бабочки) |
||||
|
Внутриутробная гипотрофия |
||||
|
Особенностистроения лицевого черепа |
||||
|
Изменения со стороны почек |
||||
|
Дополнительные критерии: |
||||
Нарушения полового развития и высокий голос |
||||
Нейроваскулярные нарушения |
||||
|
Нарушения умственного развития |
Гипоплазия является гистологическим признаком, который оценивается на основании определения отношения желчных протоков к портальным трактам менее 0,6. В норме это отношение должно быть более 0,9.
Выявление гипоплазии внутрипеченочных желчных протоков наряду с отсутствием характерного для синдрома Алажиля симптомокомплекса может свидетельствовать о несиндромальной ее форме. Однако гипоплазия может быть одним из проявлений целого ряда заболеваний и, в том числе хромосомных нарушений (например, синдром Эдвардса), инфекционных заболеваний и некоторых метаболических нарушений. Это диктует необходимость исключения данных заболеваний перед установлением диагноза несиндромальной формы гипоплазии внутрипеченочных желчных протоков.
Атрезия внепеченочных желчных протоков является наиболее частой причиной не онатального холестаза. Патогенез ее можно условно разделить на 2 этапа, первый из которых включает механизмы компенсации, направленные на «сохранение» гепатоцитов. На этом этапе отмечается снижение активности транспортных систем синусоидальной мембраны гепатоцита, что приводит к уменьшению захвата ЖК и других компонентов желчи клетками печени. Отмечается значительное увеличение проницаемости межклеточных соединений, что способствует поступлению желчи из внутрипеченочных желчных протоков в кровь. Изменяется направление внутриклеточного транспорта компонентов желчи и увеличивается проницаемость синусоидальной мембраны гепатоцита для обратного тока желчи из клеток печени в кровь. На втором этапе происходит разрушение внутрипеченочных желчных протоков с формированием характерных ступенчатых некрозов и ихпролиферации, а также деструктивные изменения гепатоцитов с формированием билиарного цирроза печени. Во внутриутробном периоде функцию экскреции и печеночно-кишечной циркуляции выполняет материнский организм, в связи с чем дети с атрезией внепеченочных желчных протоков в большинстве случаев рождаются доношенными и не имеют патологических изменений при рождении.
Первым клиническим признаком является ахолия стула, однако следует помнить о возможности отхождения мекония и, следовательно, появление обесцвеченного стула лишь на 4-5 сутки жизни. Желтуха появляется на 2-3 день жизни, а к началу второй недели кожные проявления уменьшаются. Примерно у 66% больных отмечается развитие «светлого промежутка» в течение 2-х недель. Затем желтуха вновь появляется или нарастает, приобретая зеленоватый оттенок. Характерно увеличение размеров печени к концу 1 месяца жизни, хотя уплотнение ее консистенции может быть выявлено раньше. В более поздние сроки отмечается значительное увеличение и уплотнение печени, постоянная ахолия стула, появляются признаки портальной гипертензии, спленомегалия, кожный зуд и ксантомы. Диагностическое значение имеет сочетание характерных клинических признаков с отсутствием визуализации желчного пузыря при УЗИ. Дополнительное диагностическое значение имеют результаты пункционной биопсии печени и гепатобилиарной сцинтиграфии. Гистологическое исследование биоптата печени выявляет некротически-воспалительную деструкцию как внепеченочных так и внутрипеченочных желчных протоков, признаки билирубино- и желчестаза. Могут выявляться единичные гигантские и полинуклеарные клетки, очаги экстрамедуллярного кроветворения. В последствие отмечается формирование холестаза, портального и перипортального фиброза и цирроза (рис. 8)

Рис. 8. Гистологическое исследование биоптата печени при атрезии внепеченочных желчных протоков.
Киста общего желчного протока в 2-5% случаев вызывает полное нарушение проходимости желчевыводящей системы и, следовательно, может быть причиной внепеченочного холестаза. В большинстве случаев киста сочетается с атрезией внепеченочных желчных протоков. Клинико-лабораторные проявления не отличаются от таковых при атрезии. Диагностическое значение имеет УЗИ, выявляющее полостное образование в проекции общего желчного протока.
Таким образом, развитие синдрома холестаза у новорожденных детей может быть следствием широкого спектра причин с локализацией первичного патогенетического дефекта на разных уровнях гепатобилиарной системы. Патогенетические механизмы различных заболеваний отражают характерный клинико-лабораторный симптомокомплекс, имеющий важное значение в диагностике. Независимо от причины нарушение оттока желчи приводит к реализации токсических эффектов различных ее компонентов и вовлечению в патологический процесс других органов и систем.
10 мая 2012 года в Казани состоялась республиканская научно-практическая конференция «Синдром холестаза у детей». Непосредственным организатором конференции выступила кафедра госпитальной педиатрии КГМУ (заведующий кафедрой, профессор В.П. Булатов). Для участия в конференции были приглашены многочисленные педиатры республики Татарстан, неонатологи, врачи смежных специальностей – всего порядка 150 человек. С приветствиями к аудитории обратились главный педиатр МЗ РТ – И.Г. Чигвинцева, профессор А.П. Киясов – проректор по научной и инновационной работе КГМУ, главный врач Детской республиканской клинической больницы МЗ РТ Р.Ф. Шавалиев.
 По словам главного педиатра республики Ирины Григорьевны Чигвинцевой
в последние 20 лет очень многое изменилось в диагностике и лечении заболеваний желчевыводящих путей и печени. «Во многом наш клинический опыт, расширение спектра диагностических исследований позволили выделить из общего класса заболеваний различные нозологии. Синдром холестаза как раз является одним из них. Он встречается достаточно часто среди новорожденных и связан с тяжелыми осложнениями пренатального периода, является следствием врожденной несостоятельности гепатобилиарной системы. В наших условиях, когда мы перешли к выхаживанию детей с экстремально низкой массой тела, проблемы диагностики и своевременное лечение приобретают для нас серьезный актуальный смысл».
По словам главного педиатра республики Ирины Григорьевны Чигвинцевой
в последние 20 лет очень многое изменилось в диагностике и лечении заболеваний желчевыводящих путей и печени. «Во многом наш клинический опыт, расширение спектра диагностических исследований позволили выделить из общего класса заболеваний различные нозологии. Синдром холестаза как раз является одним из них. Он встречается достаточно часто среди новорожденных и связан с тяжелыми осложнениями пренатального периода, является следствием врожденной несостоятельности гепатобилиарной системы. В наших условиях, когда мы перешли к выхаживанию детей с экстремально низкой массой тела, проблемы диагностики и своевременное лечение приобретают для нас серьезный актуальный смысл».
« У новорожденных и детей первых дней жизни синдром холестаза является одним из ранних проявлений заболеваний печени и желчных протоков. Основной предпосылкой являются морфо-функциональные особенности гепатобилиарной системы в этом возрасте. В этом возрасте отмечается относительно низкое количество аминокислот, а также незрелость всех этапов печеночных и кишечных циркуляций. Морфо-функциональные особенности предполагают развитие патологических состояний при определенных условиях», отметила профессор. Под синдромом холестаза принято понимать нарушение образований экскреции желчи по желчевыводящей системе, приводящей к повышению компонентов желчи в крови и недостаточному поступлению в кишечнике. Синдром холестаза имеет характерные клинические и лабораторные проявления. В большинстве случаев синдром холестаза сочетается с синдромом цитолиза повышения активности ферментов трансаминаз, но также и с признаками печеночно-клеточной недостаточности. Клиническими проявлениями холестаза является желтуха, гепатомегалия, ахолия стула, насыщенный цвет мочи. Тактика лечения детей с синдромом холестаза предполагает выявление факторов, способствующих развитию холестаза, затем необходимо исключение заболеваний печени, эффективность лечения которых зависит от сроков его проведения. После этого необходимо адекватное лечение основного заболевания, исключение или ограничение гепатотоксичных лекарств и препаратов крови, максимально раннее начало энтерального питания, также необходимо введение лечебного питания с повышенным содержанием СЦТ, жирорастворимых витаминов через 10 дней, урсодезоксихолиевой кислоты, противопоказано введение гормональных препаратов. По словам профессора, все заболевания желчевыводящих путей принято делить на внепеченочный холестаз и внутрипеченочный холестаз. При внутрипеченочном холестазе визуализируется ахолия стула, у внепеченочного – наоборот. В сообщении докладчик также подробно остановилась на синдроме Байлера. По словам докладчика, дети с таким синдромом составляют группу риска по развитию онкологических заболеваний. Тактика ведения детей с ПВСХ 1 и 2 типов (болезнь Байлера): информирование семьи о том, что возможен риск развития злокачественных новообразований, а также рождения больного ребенка при повторных беременностях. Также, детей с синдромом Байлера необходимо наблюдать один раз в течение двух-трех месяцев. Поддерживающая терапия будет состоять из лечебного питания, использования жирорастворимых витаминов, микро- и макроэлементов, препаратов, уменьшающих кожный зуд. Профессор в рамках сообщения упомянула и о ведении детей с синдромом Алажилль. Прогноз жизни при синдроме Алажилль зависит от течения заболевания, попытки хирургической коррекции ошибочны. Анна Владимировна подробно остановилась и на вопросе диагностики и лечения неонатального гепатита – воспалении печени, вызванном гепатотропными возбудителями: оппортунистическими чаще всего. Участникам конференции были представлены и метаболические нарушения. К ним относятся: тирозинемия, фруктоземия, нарушения синтеза желчных кислот, прогрессирующий семейный внутрипеченочный холестаз, муковисцидоз и другое. Заподозрить такие метаболические нарушения можно, если в семейном анализе присутствуют случаи смерти предыдущих детей от сепсиса с неустановленным возбудителем, синдрома внезапной смерти, неустановленной причины. Помимо этого симптомами метаболических нарушений являются нарушения общего состояния: срыгивания, рвота, потеря веса.
Далее с сообщением выступил заведующий кафедрой госпитальной терапии КГМУ, профессор В.П. Булатову. Он представил доклад «Желчнокаменная болезнь в детском возрасте: дифференцированный подход к терапии». «Желчнокаменная болезнь (ЖКБ) – это болезнь цивилизации», начал свое выступление профессор. Согласно сообщению, частота желчнокаменной болезни (холелитиаза) возросла среди лиц молодого возраста 16-35 лет, при УЗИ новорожденных первых четырех дней желчные камни обнаруживаются в 0,5%. Увеличение роста холелитиаза объясняется улучшением диагностики, а истинный рост заболевания связан с особенностями питания, гиподинамией, ухудшением экологической обстановки. Причинами образования холестериновых желчных камней являются снижение сократительной функции желчного пузыря, перенасыщение желчи холестерином. По словам профессора, билиарный сладж (инородное скопление желчи) формируется у женщин, принимающих оральные контрацептивы и у беременных, а также пациентов, находящихся на парентералом питании или у голодающих. Также к факторам формирования билиарного сладжа относится использование низкокалорийных диет с целью снижения массы тела, а также такие заболевания как: микроцитарная и серповидно-клеточная анемия, цирроз печени, вирусные гепатиты. С 1997 по 2012 год в Детской республиканской клинической больнице МЗ РТ было пролечено 99 больных с ЖКБ, преобладающее число среди них девочки в возрасте 12-14 лет. У 30% больных отмечалась наследственная предрасположенность в основном по материнской линии. Таким больным рекомендуется диета – стол №5. «Это 50% эффективного лечения», отметил профессор. В своем сообщении профессор также выделил показания к консервативной терапии при второй стадии ЖКБ. При обнаружении желчных камней и без отсутствия желчной колики у детей до трех лет рекомендуется только наблюдение. В возрасте от трех до двенадцати лет – операция показана. У детей в подростковом возрасте рекомендуется только выжидательная тактика.
В рамках конференции специалисты также обсудили такие темы как «Синдром холестаза при фетальных гепатитах», были проанализированы причины синдрома холестаза у детей, а также лечение детей с билиарной атрезией.
В завершении конференции участники обменялись мнениями и задали свои вопросы лекторам.
Альфия Хасанова
-- [ Страница 5 ] --
Наиболее значимым показателем мы считаем уровень билирубина, нормализация которого является прогностически благоприятным признаком, свидетельствующим о высокой вероятности легкого варианта течения болезни и отсутствии показаний к ТП в течение всей жизни. Независимо от варианта течения, формирование цирроза печени не типично для синдрома Алажиля, тогда как показанием к ТП являются осложнения, длительно сохраняющегося холестаза, значительно нарушающие качество жизни ребенка. Следует особенно подчеркнуть, что любое хирургическое вмешательство на желчевыводящей системе при данном синдроме значительно ухудшает состояние ребенка и способствует формированию билиарного цирроза, что показано у 3-х детей в нашем исследовании.
Динамически обследовано также 15 детей с болезнью Байлера в возрасте от 10 дней жизни до 6 лет. Болезнь Байлера всегда является показанием к ТП. Оптимальные сроки ее проведения определяются появлением патологических состояний, нарушающих качество жизни или признаков билиарного цирроза. В нашем исследовании сроки проведения ТП варьировали от 9 месяцев до 8 лет. Во всех случаях своевременно определены показания и эффективно выполнена данная операция. Установлен высокий риск развития онкологических заболеваний гепатобилиарной системы, которые были выявлены у 4 детей (27%), в среднем в возрасте 2,2+0,5 лет: печеночно-клеточная карцинома –у 3-х и холангиокарцинома – у одного больного. Полученные результаты определили необходимость рекомендовать УЗИ мониторинг и динамическое определение уровня а-ФП в процессе наблюдения детей с болезнью Байлера.
С целью выявления патогномоничных морфологических изменений и определения показаний к проведению биопсии печени изучены результаты гистологического исследования при различных заболеваниях гепатобилиарной системы. У детей с БА установлены различной степени выраженности холестаз, пролиферация желчных протоков и фиброз (таб 4).
Таб 4. Основные морфологические изменения печени у детей с билиарной атрезией в зависимости от возраста.
| Гистологические признаки | Дети с билиарной атрезией | |||
| 1,0+0,5 мес. жизни n-10 | 2,0+0,5 мес. жизни n-9 | 3,0+0,5 мес. жизни n-11 | ||
| Активность воспаления по Кнодель (баллы) | 3,7+1,5 | 2,8+1,5* | 4,1+2,4* | |
| Перипортальные и мостовидные некрозы | 8/10 | 6/9 | 7/11 | |
| Внутридольковая дегенерация и очаговые некрозы | 8/10 | 7/9 | 8/11 | |
| Портальное воспаление | 10/10 | 9/9 | 11/11 | |
| Степень выраженности фиброза по Desmet, баллы | 1,7+0,4 | 1,8+0,5* | 2,6+1,1* | |
| Порто-портальный фиброз | 6/101 | 7/92 | 5/11 | |
| Фиброзные септы | 1/10 | 1/9 | 4/11 | |
| Цирроз | 0/10 | 0/9 | 2/11 | |
| Пролиферация желчных протоков | 10/10 | 9/9 | 11/11 | |
| Степень выраженности холестаза: | ||||
| Внутриклеточный | 10/10 | 9/9 | 11/11 | |
| В желчных капилярах | 8/10 | 9/9 | 6/11 | |
| В желчных протоках | 8/10 | 7/9 | 7/11 | |
* р < 0,05, 1 – у 3-х детей 1 группы и 2 - у 1-го пациента 2 группы гистологические признаки фиброза в препаратах отсутствовали.
У всех отмечались воспалительные изменения низкой или минимальной степени активности по Кнодель. Степень выраженности холестаза и пролиферации желчных протоков была минимально выражена в 1 месяц и значительно нарастала к 3 месяцам жизни. Аналогичные закономерности выявлены в динамике формирования фиброза. Выявлена прямая зависимость между степенью выраженности морфологических изменений и возрастом ребенка (r-0,92, p<0,05). Суммарная активность воспаления по Кнодель у детей гр.1 была выше по сравнению с больными гр. 2 и ниже чем у детей гр 3, однако статистически значимых отличий не выявлено. У детей гр.2 степень выраженности воспаления была достоверно ниже, чем у детей гр.3 (р<0,05). Вместе с тем, следует отметить невысокую суммарную оценку воспаления по Кнодель у всех обследованных больных, соответствующую низкой или очень низкой гистологической степени активности.
Выявлена обратная зависимость между активностью сывороточного уровня фермента ГГТ и степенью выраженности фиброза по шкале Десмет у детей старше 1 месяца жизни (r-0,93, p<0,05), что позволяет говорить о диагностической ценности данного биохимического показателя (рис 4).
Рис.4. Взаимосвязь между степенью выраженности фиброза (в баллах по Десмет) и сывороточным уровнем фермента ГГТ у детей с билиарной атрезией (r -0,93, p<0,05).
Повышение ГГТ менее чем в 10 раз с высокой вероятностью свидетельствует о значительной степени (3-4 балла по шкале Десмет) выраженности фибротических изменений в ткани печени. Других клинико-лабораторных показателей, достоверно отражающих степень морфологических изменений при БА, выявить не удалось. При заболеваниях, проявляющихся внутрипеченочным холестазом, с целью уточнения диагноза морфологическое исследование биоптата печени проведено 35 детям в возрасте от 1 до 4 месяцев жизни. Из них с болезнью Байлера 9 детей, с синдромом Алажиля -11, с несиндромальной формой гипоплазии желчных протоков 4 ребенка, с ПСВХ 3 типа - 4, с дефицитом а-1-АТ 3 ребенка, с неонатальным гемохроматозом двое и с галактоземией двое. У всех детей выявлен холестаз, минимальная или низкая степень активности воспаления по Kнодель и фибротические изменения. Максимальная степень выраженности фиброза описана при 3 типе ПСВХ (3,75+0,5 баллов), достоверно отличающаяся от детей с болезнью Байлера (1,5+0,5 баллов) и синдромом Алажиля (1,3+0,5 баллов) (р<0,05). Характерным гистологическим признаком синдрома Алажиля является гипоплазия внутрипеченочных желчных протоков. Во всех исследованиях отношение внутрипеченочных желчных протоков к портальным трактам было меньше 0,6. Кроме того, единичные желчные протоки, которые определялись в препаратах, в большинстве случаев оказывались аномальными (рис 5). Типичным для болезни Байлера является внутриклеточное скопление крупных гранул желчи «желчи Байлера» наряду со значительным увеличением размеров гепатоцитов, выявленное у всех детей (рис 5). Характерным проявлением неонатального гемохроматоза служит внутриклеточное скопление железа (рис 5), дефицита а-1-АТ - ПАС-позитивные включения.
а
б
 в
в

Окр. по Ван-гизону Окр. гематоксилином и эозином, Окр. по Перлсу
Рис 5. Аномальный желчный проток при синдроме Алажиля (а), внутриклеточное скопление глыбок желчи – «желчь Байлер» при болезне Байлера (б), Внутриклеточное скопление железа при неонатальном гемохроматозе (в).
С целью изучения роли вирусных инфекций в генезе заболеваний гепатобилиарной системы нами проведено исследование ДНК вирусов семейства Герпес и вирусов гепатита В и С методом ПЦР в биоптате печени и в крови. Параллельно проводилось гистологическое исследование биоптата печени. В исследование включен больной с генерализованной ЦМВ инфекцией, одним из проявлений которой был гепатит. В биоптате печени ДНК ЦМВ выявлена у большинства детей с БА и у больного с генерализованной формой ЦМВИ (таб 5.). При других заболеваниях, имеющих известный этиопатогенез, данный вирус выявлен только у 2 детей.
Таб 5. Частота выявления ДНК ЦМВ в биопатате печени детей с заболеваниями гепатобилиарной системы.
| Диагноз | Биоптат печени | кровь |
| БА | 27 / 30 | 5 / 30 |
| ПСВХ 1-3 типов | 1 / 7 | 0 / 7 |
| Синдром Алажиля | 1 / 5 | 0 / 5 |
| ПСХ | 0 / 2 | 0 / 2 |
| Неонатальный гемохроматоз | 0 / 2 | 0 / 2 |
| Другие | 0 / 3 | 0 / 3 |
| ЦМВ гепатит. | 1 / 1 | 1 / 1 |
Среди разнообразных заболеваний печени наследственным пигментным гепатозам, именуемым также функциональными гипербилирубинемиями, принадлежит довольно скромное место. Клиническое значение этих болезней (или синдромов) состоит прежде всего в том, что они часто не распознаются врачами, многократно обследуются и длительное время без достаточных оснований активно лечатся, создавая иллюзию серьезности страдания.
Не случайно известный немецкий патоморфолог Н. Thaler высказался в том смысле, что при наследственно детерминированных функциональных гипербилирубинемиях опасность для пациентов исходит чаще всего от врачей, которые, впервые обнаружив желтуху, трактуют ее ошибочно, как хронический гепатит различной этиологии, и назначают многочисленные лабораторно-инструментальные исследования, а также массированную фармакотерапию, в которой они большей частью не нуждаются. E. Meulengracht считал, что «основным риском у пациентов с функциональной гипербилирубинемией является переоценка заболевания».
Определение
Пигментные гепатозы - это группа генетически детерминированных энзимопатий, клинически проявляющихся изолированным нарушением внутрипеченочного обмена билирубина с развитием хронической или перемежающейся желтухи без отчетливых изменений структуры и функции печени и явных признаков повышенного гемолиза.
Образование и обмен билирубина
Свободный (неконъюгированный) билирубин образуется при распаде гемоглобина эритроцитов и разрушении гема в ретикулоэндотелиальной системе печени, селезенки и костного мозга.
Ежесуточно наблюдается распад примерно 1% циркулирующих в крови эритроцитов и образование 250 мг свободного билирубина, который высокотоксичен и плохо растворим в воде. Поступив в общий кровоток, свободный билирубин циркулирует в крови в комплексе с альбумином, поступает в воротную вену и доставляется в печень.Роль печени в обмене билирубина состоит в захвате свободного билирубина гепатоцитами из синусоидов и перемещении через цито-плазматическую мембрану внутрь гепатоцита с помощью транспортных белков-переносчиков. Этот механизм получил наименование «flip-flap» («качели»). Сначала происходит адгезия свободного билирубина к липидной мембране гепатоцита с последующей диффузией его через билипидный слой в цитоплазму печеночной клетки.
Захват свободного билирубина в синусоидах печени, его высвобождение из комплекса с альбумином и перемещение (транслокация) в гепатоцит происходят при участии фермента билитранслоказы, локализованного в мембране эндоплазматической сети, и двух фракций неспецифических цитоплазматических белков, обозначаемых как Y- и Z-протеины (лигандины).
Они связывают холефильные органические анионы, к которым принадлежит и свободный билирубин.
В гепатоците свободный билирубин с помощью белков-переносчиков (глутатион-S-трансферазы) доставляется к мембране эндоплазматической сети, где происходит его конъюгация с одной или двумя молекулами глюкуроновой кислоты и образование связанного билирубина (моно- и диглюкуронида в соотношении 1:3). Этот процесс катализирует фермент уридиндифосфатглюкуронилтрансфераза. В последнее время установлено, что процесс глюкуронизации свободного билирубина осуществляется в основном с помощью микросомального изофермента - УДФ-ГТ 1А1, который превращает неполярные жирорастворимые субстраты в водорастворимые. Последние, в свою очередь, выводятся из организма с желчью и мочой.
Образовавшийся в микросомальной системе фермента УДФ-ГТ конъюгированный (связанный) билирубин отличается от свободного билирубина низкой токсичностью и является водорастворимым соединением.
В этом, в сущности, состоит биологический смысл реакции конъюгации свободного билирубина. Важно отметить, что этот процесс происходит однонаправленно - от синусоидального к билиарному полюсу гепатоцита.Не менее сложен и не до конца расшифрован процесс перемещения связанного билирубина (водорастворимого конъюгата билирубина) в первичные желчные канальцы. Это - конечный этап обмена билирубина в печеночной клетке. Транслокация конъюгированного билирубина из гепатоцита в желчные канальцы происходит при участии донатора энергии - АТФ-зависимой транспортной системы. На этот процесс оказывают влияние: градиент концентрации; цитоплазмати-ческая мембрана билиарного полюса гепатоцита; лизосомы; пластинчатый комплекс (аппарат Гольджи) и скорость секреции желчи.
В желчи образуется макромолекулярный (липидный) комплекс, выполняющий роль транспортной системы (в виде сложных мицелл и везикул), который обеспечивает перемещение всех компонентов желчи по желчным протокам и сохранение ее коллоидной стабильности. Помимо связанного билирубина, в состав макромолекулярного комплекса входят: холестерин, фосфолипиды, соли желчных кислот, белки и др.
В кишечнике под влиянием бактериальной флоры, вырабатывающей ферменты (дегидрогеназы), связанный билирубин восстанавливается до уробилиногена (бесцветное соединение), который частично всасывается в дистальных отделах тонкой кишки и по воротной вене возвращается в печень (кишечно-печеночная циркуляция уробилиногена), где разрушается до дипирролов. Большая часть уробилиногена поступает в толстую кишку и выделяется с калом в виде фекального уробилиногена (стеркобилиногена) в количестве 47-276 мг/сут. Билирубинурия появляется только при избыточном накоплении в крови связанного билирубина.
Классификация
В настоящее время в группу наследственных пигментных гепато-зов («функциональных гипербилирубинемий») включают 8 ее клини-ко-патогенетических форм.
С неконъюгированной гипербилирубинемией.
1. Синдром Жильбера (и его вариант: постгепатитную гиперби-лирубинемию).
2. Синдром (болезнь) Мейленграхта.
3. Синдром Криглера-Найара I и II типа.
4. Синдром Люси-Дрисколла.
С конъюгированной гипербилирубинемией.
1. Синдром Дабина-Джонсона.
2. Синдром Ротора.
3. Болезнь Байлера.
4. Синдром Аагенеса-Саммерскилла.
Две последние формы (болезнь Байлера и синдром Аагенеса) относятся к врожденным внутрипеченочным холестазам. Еще недавно наличие холестаза служило основанием для исключения из группы «функциональных гипербилирубинемий». В последнее время их стали рассматривать, как представителей семейных гипербилируби-немий.
Синдром Жильбера
Синдром Жильбера - самая распространенная форма функциональных гипербилирубинемий: в различных регионах мира встречается с частотой от 1-5 до 11-12% в популяции.
Первое упоминание о синдроме Жильбера связано с именем A. Gilbert et al. , которые представили его подробное описание под названием «простая семейная холемия» (cholemia simple familiale). В последующие годы синдром Жильбера именовали по-разному: «идиопатическая неконъюгированная гипербилирубинемия»; «семейная негемолитическая желтуха»; «семейная перемежающаяся желтуха»; «хронический доброкачественный пигментный гепатоз» и др.
В основе синдрома Жильбера лежит наследственно детерминированный генный дефект микросомального фермента УДФ-ГТ, обусловливающий частичную редукцию печеночного клиренса свободного (неконъюгированного) билирубина и накопление его в крови. В промоторном участке (регионе) А (ТА)6 ТАА-гена, кодирующего микросомальный фермент УДФ-ГТ имеется дополнительный динуклеотид ТА, который вызывает образование участка (региона) А (ТА)7 ТАА. Это приводит к снижению активности фермента УДФ-ГТ 1А1, ответственного за конъюгацию свободного билирубина с глюкуроновой кислотой и образование связанного билирубина. Этот процесс снижается до 30% от нормы. Кроме того, при синдроме Жильбера установлен дефицит фермента билитранс-феразы и Y- и Z-белков (их сейчас идентифицируют с ферментом глутатион-S-трансферазой), в связи в чем нарушается захват (экстракция) свободного билирубина из плазмы крови в синусоидах печени, перенос его в цитоплазму гепатоцита и транспортировка к микросомам печеночной клетки. Это приводит к избыточному накоплению в крови свободного билирубина.
Нет единого мнения в вопросе о типе наследования при синдроме Жильбера. В последнее время склоняются к аутосомно-доминантному типу наследования, но с неполной пенетрантностью, т. е. с различной частотой проявления дефектного гена в фенотипе его носителей.
Таким образом, синдром Жильбера - это, по-видимому, не болезнь, а особое состояние (sui generis), обусловленное врожденным дефектом - дефицитом микросомального фермента УДФ-ГТ.
Синдром Жильбера манифестирует обычно в подростковом, юношеском или молодом возрасте (от 7 до 28-30 лет), причем чаще выявляется у мужчин (в соотношении 3-7:1). Тот факт, что синдром Жильбера проявляет себя чаще всего в периоде полового созревания у мужчин, возможно, указывает на определенную роль в клиренсе билирубина мужских половых гормонов (андрогенов).
У значительной части пациентов синдром Жильбера длительное время протекает латентно или субклинически, поэтому нередко его обнаруживают случайно. Например, при биохимическом анализе крови определяют повышенный уровень свободного билирубина или при обследовании больных по поводу других заболеваний выявляют субикте-ричность склер и легкую желтушную окраску кожных покровов и т. п.
Для синдрома Жильбера характерны: матово-желтая окраска кожи лица, носогуб-ного треугольника и подмышечных впадин; гиперпигментация кожи в окружности глаз. А. Жильбер описал типичную «диагностическую триаду» признаков:
печеночная «маска» (желтушность);
ксантелазмы на веках;
волнообразность появления и исчезновения симптомов.
Отмечают, что пигментация кожи нарастает под влиянием световых лучей и тепла, химических и механических раздражителей. Примерно у 50% пациентов с синдромом Жильбера наблюдается клиническая симптоматика: тупые боли или чувство тяжести в правом подреберье, диспепсические явления (понижение аппетита, тошнота, запор или диарея и др.); зябкость с появлением «гусиной кожи»; мигренеподобная головная боль; склонность к брадикардии и артериальной гипо-тензии; нейромышечная перевозбудимость. Часто при синдроме Жильбера определяется астеновегетативный синдром, повышенная тревожность, депрессия или легкая возбудимость, нарушения ночного сна, биоритмологические сдвиги. У 15-20% пациентов печень слегка увеличена (на 1-2 см), безболезненна, обычной консистенции. Иногда выявляют дисфункцию желчного пузыря и сфинктерного аппарата внепеченочных желчных путей.
Важно подчеркнуть, что появление клинических симптомов при синдроме Жильбера, в том числе нарастание желтухи (гипербилирубинемии) нередко провоцируется интеркуррентной инфекцией, голоданием, психическими и физическими перегрузками, алкоголем.
Можно удивляться наблюдательности М.Ю. Лермонтова, который в романе «Герой нашего времени» (повесть «Княжна Мери») описал появление характерных признаков синдрома Жильбера у Печорина, которые развились у него на фоне тяжелых психических переживаний: «Я вернулся домой... Ядовитая злость мало-помалу заполняла мою душу. Я не спал всю ночь. К утру я был желт как померанец» (вид апельсина). Следует напомнить, что Печорин был молод, импульсивен, эмоционально лабилен, но обладал крепким физическим здоровьем. В общем анализе крови при синдроме Жильбера, как правило, нет анемии, ретикулоцитоза; снижения осмотической стойкости эритроцитов и продолжительности их жизни (нет признаков гемолиза); скорость оседания эритроцитов - в пределах нормы; изредка наблюдается повышенный уровень гемоглобина (до 150 г/л).
В биохимическом анализе крови отсутствуют признаки цитолиза, холестаза, гепатоцеллюлярной недостаточности (уровень аминотрансфераз, ЩФ, у-ГТП, содержание холестерина и фосфолипидов, альбуминов остаются в норме). Не определяется билирубинурия.
Специальные методы диагностики.
Проба с бромсульфалеином (Caroli): после внутривенного введения 5% раствора бромсульфалеина (из расчета 5 нг/кг массы тела) определяют время его появления в дуоденальном содержимом. Для этого каждые 30 с помещают каплю содержимого двенадцатиперстной кишки в 10 N раствор едкого натрия - на присутствие бромсульфалеина указывает фиолетовое окрашивание (хромодиагностика). При синдроме Жильбера отмечается задержка элиминации индикатора до 20-40 мин (в норме 5-15 мин). Можно также определять элиминацию бромсульфалеина РЭС печени. С этой целью до и через 45 мин после внутривенного вливания бромсульфалеина определяют содержание индикатора в крови. При синдроме Жильбера в кровяном русле остается >10% введенного красителя (в норме
Проба с бенгалроз-13 (радионуклидный метод определения поглотительно-экскреторной функции печени). При синдроме Жильбера полупериод клиренса индикатора, меченного радионуклидом, удлиняется с 13 до 28 мин, а время максимального накопления увеличивается с 1,5 до 4,2 ч.
Проба с никотиновой кислотой. Никотиновую кислоту вводят внутривенно в дозе 50 мг или принимают внутрь 170 мг утром натощак. До ее введения и через 3 ч после «нагрузки» никотиновой кислотой определяют уровень свободного билирубина. При синдроме Жильбера он возрастает в 2 раза и более, в основном за счет билирубинмоноглюкуронида (в норме преобладает билирубиндиглюкуронид).
Проба с гипокалорийной диетой (400 кКал/сут): через 24-48 ч содержание свободного билирубина в крови увеличивается в 1,5-2 раза (до 30-50 мкмоль/л).
Тест с рифампицином (Vesilla, 1993): прием 900 мг рифампицина вызывает повышение уровня неконъюгированного билирубина в крови в 1,5 раза (аналогичный эффект получен также при введении анаболических стероидов).
Проба с фенобарбиталом: прием препарата, являющегося индуктором (активатором) микросомального фермента УДФ-ГТ, в дозе 3 мг/кг массы тела в сутки в течение 5 дней существенно снижает содержание свободного билирубина в крови (диагноз ex juvantibus).
Отдельные авторы отмечают, что при синдроме Жильбера повышено выделение с мочой копропорфиринов, в основном за счет изомера 1-го типа (50-80%). Морфологически (биопсия) при синдроме Жильбера структура печени, как правило, не изменена, но в центре печеночных долек у билиарного полюса гепатоцитов наблюдается характерное накопление пылевидного золотисто-коричневого пигмента, не содержащего железа, - липофусцина. Предположительно, липофусцин образуется в результате реакции аутооксидации металлофлавопротеидов. Выполняя адаптационную функцию, липофусцин служит дополнительным источником энергии для гепатоцитов.
В Пермском гастроцентре мы наблюдали в течение нескольких лет 76 пациентов с синдромом Жильбера. Группа комплектовалась постепенно: первоначально пациенты с синдромом Жильбера отбирались из числа лиц, направлявшихся участковыми терапевтами на консультацию по поводу субиктерич-ности склер и кожных покровов неясного происхождения или с предположительным диагнозом хронического гепатита или холецистита. После всестороннего обследования и исключения вирусных, алкогольных, холестатических заболеваний печени, а также проведения специальных диагностических проб (тестов) с нагрузкой никотиновой кислотой, с бенгалроз-131 , с гипокалорийной диетой, с фенобарбиталом устанавливали диагноз синдрома Жильбера. В сомнительных случаях (редко) проводили биопсию печени и микроскопическое изучение биопсий-ного материала. В дальнейшем комплектование и пополнение группы с синдромом Жильбера происходило после приглашения в гастроцентр и всестороннего обследования их кровных родственников первой степени родства. Как известно, синдроме Жильбера передается по наследству, чаще по материнской линии. Таким путем нам удалось обнаружить целые семьи, где диагноз синдрома Жильбера был установлен у 2-3 и даже у 6 членов одной семьи (мать, 2 сына, ее брат и 2 сестры). Среди них оказалось немало лиц с латентным течением синдроме Жильбера. Часть пациентов с синдромом Жильбера предъявляла жалобы на общую слабость, утомляемость, субиктеричность склер и кожных покровов; раздражительность, эмоциональную лабильность; головные боли, слезливость. Не было кожного зуда, телеангиэктазий и пальмарной эритемы.
Можно предположить, что часть невротических жалоб при синдроме Жильбера имеет ятрогенное происхождение, поскольку врачи, почти как правило, не разъясняют пациентам наследственный и доброкачественный характер желтухи при синдроме Жильбера, что может стать причиной для беспокойства и тревоги за состояние своего здоровья, особенно у мнительных лиц. В 39,3% случаев появление или нарастание субиктеричности склер и кожных покровов провоцировалось интеркуррентными инфекциями, у 13% - психическими или физическими перегрузками (у одного из школьников желтуха появилась после лыжного кросса, проводившегося без предварительных тренировок, причем уровень свободного билирубина повысился в 3 раза). В 80% случаев желтуха имела интермиттирующий характер, в 20% была постоянной. Функциональные пробы печени у всех пациентов с синдромом Жильбера были в пределах нормы. УЗИ и компьютерная томография, а также пункционная биопсия, проводившаяся в сомнительных случаях (у двух человек), не выявили структурных изменений в печени. Уровень общего билирубина варьировал от 25 до 65 мкмоль/л, в том числе свободного - от 22 до 59, а связанного - от 5,1 до 8 мкмоль/л.
Постгепатитная гипербилирубинемия развивается у 2-4% больных, перенесших острый вирусный гепатит. По современным воззрениям, постгепатитная гипербилирубинемия не является самостоятельной формой функциональных гипербилирубинемий, а рассматривается как вариант синдрома Жильбера. Среди 76 наблюдавшихся нами пациентов постгепатитная гипербилирубинемия была диагностирована у 1. Обычно постгепатитная гипербилирубинемия развивается в сроки от 6 мес до 2 лет после острого вирусного гепатита и достигает уровня 50 мкмоль/л. Со временем она может исчезнуть, но в части случаев приобретает хроническое интермиттирующее течение, причем уровень свободного билирубина нарастает после злоупотребления алкоголем, физических и психических перегрузок, что характерно для синдрома Жильбера. Печень может быть незначительно увеличена (1-1,5 см), но ее структура и функции не изменены. Большинство исследователей считают, что развитие неконъюгиро-ванной гипербилирубинемии у лиц, перенесших острый вирусный гепатит, - это манифестация скрыто протекавшего у них синдром Жильбера - врожденного снижения активности микросомального фермента УДФ-ГТ. В связи с этим предложения некоторых авторов выделять 2 формы синдрома Жильбера: конституциональную (наследственную) и приобретенную недостаточно обоснованы. Вместе с тем следует помнить, что в части случаев острого вирусного гепатита (В, С) может подвергнуться хронизации с развитием в дальнейшем хронического вирусного гепатита и цирроза печени.
Синдром (болезнь) Мейленграхта
Синдром (болезнь) Мейленграхта впервые описан E. Meulen-gracht в 1939 г. под названием «ювенильная перемежающаяся желтуха» (icterus juvenilis intermittens). Манифестация СМ происходит обычно в периоде полового созревания (13-17 лет) и сопровождается повышенной возбудимостью внешней нервной системы.
Одно время считали, что между синдромом Жильбера и синдромом Мейленграхта нет существенных различий и объединяли их под общим названием «синдром Жильбера- Мейленграхта». Однако при углубленном изучении патогенеза неконъюгированной гипербилирубинемии при синдроме Жильбера и синдроме Мейленграхта были установлены принципиальные различия. Так, при синдроме Жильбера снижена и активность микросомального фермента УДФ-ГТ, ответственного за синтез связанного билирубина в гепатоците, и захват свободного билирубина из крови, и его транслокация в гепатоцит, а при синдроме Мейленграхтаопределяется только врожденный дефицит фермента УДФ-ГТ, но мембрана гепатоцита активно участвует в захвате свободного билирубина из крови. Так что это все-таки две разные, хотя и родственные формы неконъюгированной гипербилирубинемии.
Синдром Мейленграхта относится к семейным гипербилирубинемиям и клинически проявляется периодически возникающей субиктеричностью склер и кожных покровов, повышенной утомляемостью, вялостью, тупыми болями в правом подреберье, диспепсическими явлениями. Возможно усиление желтушности после психических переживаний и чрезмерных физических усилий. Размеры печени и ее функции, как правило, не изменены. Повышен уровень свободного билирубина в крови (до 80 мкмоль/л), но нет признаков внутрисосудистого гемолиза эритроцитов.В значительной части случаев синдрома Жильбера, постгепатитной гипербилирубинемии и синдрома Мейленграхта не требуют медикаментозного лечения - достаточно придерживаться диетических ограничений, не употреблять алкогольные напитки, вести правильный образ жизни.
При частых эпизодах неконъюгированной гипербилирубинемии (>50 мкмоль/л), сочетающейся с клинической симптоматикой, возникает необходимость в назначении индукторов (активаторов) микросо-мального фермента УДФ-ГТ и транспортных белков - фенобарбитала в дозе 50 мг 2-3 раза в день или зиксорина - по 600 мг/сут в течение 2 нед. Обычно уже через 10 дней уровень свободного билирубина в крови снижается до нормы.
Синдром Криглера-Найяра
Синдром Криглера-Найяра впервые описан в 1952 г. J.F. Crigler и V.A. Najjar у новорожденных детей под названием «врожденная семейная негемолитическая ядерная желтуха» (congenital familial non-hemolytic jaundice with kernicterus).
Синдром Криглера-Найяра -редкая врожденная патология с аутосомно-рецессивным типом наследования. Клинические симптомы появляются у ребенка в первые часы и дни жизни и имеют неуклонно прогрессирующее течение с нарастающим повышением содержания в крови неко-нъюгированного билирубина (в 15-20 раз - до 340-680 мкмоль/л) и появлением признаков желтухи ядерного типа. При этом отсутствует несовместимость групп крови матери и ребенка.
Наследственный дефект передается только гомозиготами и локализуется в одном из 5 экзонов (1А-5) гена фермента УДФ-ГТ, несущего ответственность за конъюгацию свободного билирубина с глюкуро-новой кислотой. Дефект заключается в делеции в гене, который является общим для всех ферментов УДФ-ГТ, что обусловливает преждевременное появление стоп-кадонов и синтез дефектных (неактивных) форм УДФ-ГТ. Синтез УДФ-ГТ кодирует локус ugt-1, представляющий собой комплекс из 6 транскрипционных единиц - по 4 экзона в каждой. Различают две генетически гетерогенные формы синдрома Криглера-Найяра: I типа и II типа.
При синдроме Криглера-Найяра I типа в желчи фактически полностью отсутствует связанный билирубин. Уровень свободного билирубина в крови, как правило, превышает 200 мкмоль/л. Поскольку у новорожденных детей почти не функционирует гематоэнцефалический барьер, высокотоксичный свободный билирубин легко преодолевает его и накапливается в базальных ядрах серого вещества больших полушарий головного мозга, вызывая их повреждение и тяжелую ядерную желтуху. Проникая в клетки и митохондрии, свободный билирубин блокирует процессы окислительного фосфорилирования в гипоталамусе, хвостатом ядре, подкорковых ядрах и мозжечке с развитием «билирубино-вой энцефалопатии», которая клинически проявляется тоническими и спастическими судорогами, опистотонусом, нистагмом, атетозным гиперкинезом и мышечным гипертонусом и в итоге приводит к гибели ребенка в первые часы и дни жизни.
Некоторые авторы полагают, что в развитии неконъюгирован-ной гипербилирубинемии при синдроме Криглера-Найяра нельзя полностью исключить значения нарушений механизма образования комплекса свободного билирубина с альбуминами крови, что находит подтверждение в определенном лечебном эффекте при синдроме Криглера-Найяра растворов альбумина. Помимо неврологических симптомов, при синдроме Криглера-Найяра I типа отмечается отставание детей в физическом и психическом развитии; увеличение печени и селезенки. В общем и биохимическом анализах крови, как правило, отклонений нет. Кал ахоличен; билирубинурия и уробилинурия отсутствуют.
При синдроме Криглера-Найяра II типа в гепатоцитах синтезируется неполноценный фермент УДФ-ГТ со сниженной активностью и наблюдается мутация гена в экзонах 1А-5, но со смешанной гетерозиготностью. Тип наследования - аутосомно-рецессивный. В гепатоцитах частично образуется связанный билирубин, который поступает в желчь. Уровень свободного билирубина в крови повышен в 5-20 раз, но не превышает 200 мкмоль/л. Желчь окрашена; в кале присутствует стеркобилин. Признаки «билирубиновой энцефалопатии» обычно отсутствуют или нерезко выражены. Желтуха появляется в первые недели после рождения ребенка и имеет тенденцию к прогрессированию. Билирубинурия отсутствует; структура и функции печени сохранены. Прогноз более благоприятен, чем при синдроме Криглера-Найяра I типа.
Лечение синдрома Криглера-Найяра . Медикаментозное лечение синдрома Криглера-Найяра I типа малоперспективно; фенобарбитал неэффективен. Для лечения используют в основном частые повторные сеансы фототерапии: облучение ребенка светом с длиной волны 450 нм. Каждый сеанс длится до 16 ч/сут; при этом глаза ребенка должны быть защищены. При фототерапии происходит фотохимическое превращение высокотоксичного свободного билирубина в относительно малотоксичный и стабильный пространственный изомер - люмирубин. Эффект проявляется в снижении до 50% уровня свободного билирубина в крови, что способствует продлению жизни ребенка. Однако к 15-20 годам у многих из них все-таки развивается ядерная желтуха и возникает угроза жизни. По этой причине фототерапию следует, по-видимому, рассматривать прежде всего как подготовку к трансплантации печени, являющейся радикальным средством лечения. Побочные эффекты фототерапии: внутрисосудистый гемолиз и бронзовый цвет кожи. Испытываются также различные вспомогательные методы терапии синдрома Криглера-Найяра I типа: повторные кровопускания; обменные переливания крови; плазмаферез; внутривенные инфузии 10-20% растворов альбумина, но их эффект непродолжителен.
Лечение синдрома Криглера-Найяра II типа. Определенный эффект при синдроме Криглера-Найяра II типа может быть получен при назначении фенобарбитала, индуцирующего синтез микросомального фермента УДФ-ГТ, в дозе 30-180 мг/сут в течение 3-4 нед. У больных снижается уровень свободного билирубина в крови, улучшается их общее самочувствие. Отдельные авторы рекомендуют прием клофибрата и ипомедиола, но доказательств их эффективности мы не обнаружили. В части случаев больным синдромом Криглера-Найяра II типа также требуется фототерапия.
Синдром Люси-Дрисколла
Синдром Люси-Дрисколла - редкий вариант наследственного пигментного гепатоза, который именуют «транзиторной семейной гипербилирубинемией новорожденных» (transient familial neonatal hyperbilirubinemia). Описан в 1960 г. J.F. Lucey и Y.M. Driskoll. Синдром Люси-Дрисколла развивается уже в первые дни жизни ребенка, проявляясь интенсивной, неуклонно нарастающей желтухой ядерного типа с накоплением в крови неконъюгированного билирубина, что в части случаев может обусловить развитие «билирубиновой энцефалопатии» и летальный исход.
Установлено, что развитие синдрома Люси-Дрисколла объясняется наличием в молоке матери ингибиторов конъюгации свободного билирубина с глюкуро-новой кислотой, которые передаются ребенку при грудном вскармливании. Попадая в общий кровоток, а затем в печень, они блокируют микросомальный фермент УДФ-ГТ и синтез связанного билирубина. В настоящее время эти ингибиторы идентифицированы: они представляют собой прегнан-3в и 2Оа-диол. При своевременном установлении истинной причины развившейся желтухи и переводе ребенка на искусственное вскармливание неконъюгированная гипербилирубинемия постепенно уменьшается и исчезает; в течение 1-2 мес наступает выздоровление.
Синдром Дабина-Джонсона
Синдром Дабина-Джонсона впервые описан в 1954 г. I.N. Dubin и F.B. Johnson под названием «хроническая идиопатическая желтуха» (chronic idiopathic jaundice).
Синдром Дабина-Джонсона - наследственно детерминированная конъюгированная гипербилирубинемия с аутосомно-доминантным типом наследования. Синдром Дабина-Джонсона развивается вследствие несостоятельности АТФ-зависимой канальцевой транспортной системы, локализованной в билиарной мембране гепатоцита, что затрудняет экскрецию связанного билирубина в желчь и вызывает его частичное поступление (рефлюкс, регур-гитацию) из гепатоцита в кровь. Как установлено, в части случаев cиндрома Дабина-Джонсона наблюдается также затруднение при захвате и переносе свободного билирубина из крови в гепатоцит, поэтому гипербилирубинемия часто имеет смешанный характер: повышается одновременно содержание и связанного, и свободного билирубина. Гипербилирубинемия достигает уровня 100 мкмоль/л и выше, в основном за счет конъюги-рованной фракции. Клинически cиндром Дабина-Джонсона проявляется повышенной утомляемостью, абдоминальными болями, вплоть до коликообразных, локализующимися в правом подреберье, тошнотой, кожным зудом, иногда - диареей, а также симптомами вегетативной дистонии. В редких случаях возможен субфебрилитет. 20-30% пациентов cиндромом Дабина-Джонсона жалоб не предъявляет. В небольшой части случаев увеличены размеры печени, очень редко - селезенки. У 50% пациентов кал ахоличен, наблюдается билирубинурия, а также повышенная экскреция копропорфиринов с мочой, причем 50-80% из них составляет изомер I типа. Функциональные пробы печени, как правило, не изменены.
При визуальном осмотре через лапароскоп печень при cиндроме Дабина-Джонсона окрашена в коричнево-черный цвет (black liver disease). Морфологическое исследование биоптатов печени обнаруживает в цитоплазме гепатоцитов аморфные пигментные включения, локализованные перибилиарно, преимущественно в центре долек, которые обычно не затрагивают купферовские клетки. Они представляют собой округлые грубозернистые включения желтовато-коричневого цвета («шоколадная печень»), содержащие липофусцин (вещество из группы хромолипоидов). Их появление связывают с нарушением секреции анионных метаболитов некоторых аминокислот (тирозина, триптофана, фенилаланина). При электронной микроскопии биоптатов печени на билиарном полюсе гепатоцитов полностью отсутствуют микроворсинки или их количество резко снижено. Пероральная и внутривенная холецис-тография не выявляет тени желчного пузыря («отрицательная холецистография»), так как контрастное вещество в желчь не поступает. Однако признаков холестаза нет.
Характерны результаты бромсульфалеиновой пробы. После внутривенного введения красителя его концентрация в период с 20-й по 45-ю мин снижается, а с 90-й по 120-ю мин возрастает. Задержка индикатора в печени достигает 7-10 ч. Радионуклидная проба с бенгалроз-13 обнаруживает удлинение периода полувыведения радионуклида до 7 ч. Течение cиндрома Дабина-Джонсона волнообразное. Прогноз благоприятный. Лечение не разработано. Рекомендуют витамины, соблюдение диеты, отказ от приема алкоголя.
Синдром Ротора
Синдром Ротора впервые описан A.B. Rotor et al. в 1948 г. под названием «семейная негемолитическая конъюгированная желтуха» (familial non-hemolytic joundice with direct van den Bergh reaction).
Синдром Ротора - это редкая, наследственно детерминированная, преимущественно конъюгированная гипербилирубинемия с аутосомно-доминантным типом наследования. Манифестирует обычно с момента рождения ребенка, иногда - в пубертатном периоде, чаще у детей мужского пола. Клинически проявляется хронической или интермиттирующей нерезко выраженной желтухой. Иногда гипербилирубинемия достигает 100 мкмоль/л, главным образом за счет связанного билирубина, с преобладанием билирубинмоноглюкуронидов. У значительной части пациентов с синдромом Ротора наблюдается также умеренное повышение фракции свободного билирубина. Часть больных с синдромом Ротора предъявляет диспепсические жалобы, испытывает болевые ощущения в правом подреберье, указывает на снижение аппетита, повышенную утомляемость, однако их общее состояние нарушается мало. Возможно и бессимптомное течение синдрома Ротора. Иногда определяется небольшое увеличение размеров печени, но селезенка остается в пределах нормы. Функции печени, как правило, не нарушены. Содержание стеркобилина в кале периодически снижено, и он становится гипохоличен. В моче определяется билирубинурия; содержание копропорфиринов в моче незначительно повышено, преобладает изомер-I. Признаков гемолиза нет.
Бромсульфалеиновая проба регистрирует задержку выделения красителя до 45 мин, но отсутствует характерный для синдрома Дабина-Джонсона вторичный подъем («пик») концентрации индикатора в крови. Лапароскопия не выявляет коричнево-черной окраски печени, а при гистологическом исследовании ее биоптатов в гепатоцитах отсутствуют зерна темного пигмента, но в части случаев определяется мелкокапельная жировая дистрофия по ходу желчных канальцев.
При внутривенной холецистографии желчный пузырь не визуализируется, но пероральная холецистография выявляет тень желчного пузыря. В основе синдрома Ротора лежит врожденное ослабление АТФ-зависимой транспортной системы, специфичной для некоторых мультивалентных анионов, в том числе для связанного билирубина. Частично нарушен и транспорт свободного билирубина из крови в гепатоцит. В результате возникает затруднение экскреции связанного билирубина из гепатоцита в желчь на уровне аппарата Гольжди, в периканикулярной зоне и/или на билиарном полюсе гепатоцита с последующим рефлюк-сом (регургитацией) связанного билирубина в кровь. По своим клиническим проявлениям и механизму развития синдрома Ротора близок к синдрому Дабина-Джонсона, но не идентичен ему. Общее состояние пациентов страдает мало; прогноз благоприятен. Лечение не разработано.
Болезнь Байлера
Болезнь Байлера - болезнь Байлера (Byler disease), или прогрессирующий семейный внутрипеченочный холестаз (progressive familial intrahepatic cholestasis), была впервые описана в 1975 г. Byler - это не фамилия автора, выделившего эту нозологическую форму холестаза, а семья, принадлежащая к секте амонитов, у членов которой была впервые обнаружена эта болезнь.
Болезнь Байлера, именуемая также злокачественным семейным холестазом, - очень редкое, генетически обусловленное заболевание. Тип наследования точно не установлен, но предполагается аутосомно-рецессивный. Патологический ген локализован в хромосоме 18. Болезнь Байлера развивается в первую неделю жизни ребенка и протекает с тяжелым прогрессирующим внутрипеченочным холестазом и конъюгированной гипербили-рубинемией, достигающей 300 мкмоль/л.
Клинически проявляется нарастающей желтухой, кожным зудом; умеренной гепато- и спленомегалией, билирубинурией и выделением гипохоличного кала. В последнее время выделяют 4 клинико-патогенетических варианта болезни Байлера. В основе болезни Байлера, как полагают, лежит развитие перипортального фиброза и пролиферация желчных протоков, которые препятствуют оттоку желчи и формируют внутрипеченочный холестаз. Определенное значение в развитии болезни Байлера придают также нарушению функций микро-филаментов или канальцевой мембраны, затрудняющих экскрецию связанного билирубина из гепатоцита в желчные канальцы. Прогноз болезни Байлера неблагоприятный: летальный исход наступает обычно в возрасте до 8 лет. Медикаментозного лечения не существует. Спасение детей возможно при своевременной трансплантации печени.
Синдром Аагенеса-Саммерскилла
Синдром Аагенеса-Саммерскилла - доброкачественный возвратный внутрипеченочный семейный холестаз (benign recurrent intrahepatic familial cholestais), именуемый также «норвежским холестазом».
Синдром Аагенеса-Саммерскилла - это генетически обусловленный холестаз, имеющий в части случаев семейный характер, с аутосомно-рецессивным типом наследования. Манифестирует в неонатальном периоде, обычно в возрасте до 10 лет. В последующем (у взрослых) приобретает интермиттирующее течение с множественными эпизодами холестатической желтухи, персистирующей на протяжении 3-4 мес, которые разрешаются самостоятельно, а затем возникают вновь с интервалами от нескольких месяцев до нескольких лет. Описан случай синдрома Аагенеса-Саммерскилла, который наблюдался в течение 38 лет, причем больной за эти годы перенес 27 рецидивов холестатической желтухи и 3 лапаротомии (в связи с подозрением на механическую желтуху), при которых ничего не было обнаружено.
Клинически синдром Аагенеса-Саммерскилла протекает с желтухой, кожным зудом, иногда - с рвотой и болями в правом подреберье (у 25-50%), отсутствием аппетита; общей слабостью, похудением, гриппоподобным состоянием. Отмечается гипербилирубинемия за счет связанного билирубина, повышенный уровень холестатических ферментов при отсутствии признаков цитолиза и гепатоцеллюлярной недостаточности. Морфологически (биопсия) выявляют внутрипеченочный холестаз, желчные тромбы, расширение портальных трактов, дегенеративные изменения гепатоцитов в зоне 1. Вне рецидива функции и структура печени не изменены. В основе синдрома Аагенеса-Саммерскилла лежит дефект хромосомы 18, обусловливающий нарушение метаболизма желчных кислот и экскреции желчи, а также гуморальной регуляции этих процессов.
В качестве непосредственной причины синдрома Аагенеса-Саммерскилла называют гипоплазию лимфатических сосудов печени, которая вызывает нарушение ее функций с развитием внутрипеченочного холестаза. Указывают также на значение дефицита витамина Е, влекущего за собой развитие дегенеративных изменений в ткани печени.
Лечение синдрома Аагенеса-Саммерскилла . Рекомендуют применение глюкокортикоидов и гептрала (адеметионина), но они недостаточно эффективны. Противоречивые результаты получены при использовании препаратов урсодезоксихолевой кислоты (урсофальк, урсосан).
Спорные вопросы терминологии
Наследственные гипербилирубинемии чаще всего именуют «доброкачественными», «функциональными» или «пигментными гепатозами».
Термин «доброкачественные гипербилирубинемии» неприемлем, поскольку некоторые из этих наследственных синдромов (Криглера-Найара, болезнь Байлера, синдром Люси-Дрисколла) приводят зачастую к фатальному для больных исходу.
Не выдерживает критики и термин «функциональные гипербилирубинемии»: при углубленном гистологическом исследовании биоптатов печени методами световой и электронной микроскопии выявляют определенные морфологические изменения на клеточном и субклеточном уровнях (синдром Дабина-Джонсона, болезнь Байлера, синдром Аагенеса-Саммерскилла и др.). Известный отечественный патолог Д.С. Саркисов утверждал, что функциональных болезней и синдромов в природе вообще не существует, - все они являются структурно-функциональными, с чем нельзя не согласиться.
Наиболее удачен, по нашему мнению, термин «наследственные пигментные гепатозы», предложенный А.Ф. Блюгером, который подчеркивает печеночное происхождение гипербилирубинемий, указывает на наличие и характер морфологических изменений в печени (гепатоз), а также наследственную детерминированность синдромов и болезней, объединяемых этим термином. Еще одно частное замечание. Мы считаем нежелательным употребление терминов «прямой» и «непрямой» билирубин, которым часто подменяют термины «связанный (конъюгированный)» и «свободный (неконъюгированный)» билирубин.
«Прямого» и «непрямого» билирубина в природе не существует, - есть прямая и непрямая реакции Ван ден Берга, которые используют для определения свободной и связанной фракций билирубина. Однако помимо колориметрического метода, основанного на реакции Ван ден Берга, фракции билирубина можно определять и спектрометрическим способом, базирующемся на обнаружении характерных абсорбционных полос при 450 нм, и другими методами. Кроме того, никто почему-то до сих пор не обратил внимания на несоответствие смысловых значений терминов «свободный - непрямой» и «связанный - прямой».
Выдающийся клиницист и ученый В.Х. Василенко утверждал: «Точная терминология (claritas defitionis) характеризует уровень науки и безусловно необходима для взаимопонимания», а «отсутствие точной терминологии недостойно науки».